Congenital Muscular Dystrophy: General Overview
Author: Ian C. Langtree - Writer/Editor for Disabled World (DW)
Published: 5 Mar 2009 - Updated: 1 Feb 2023
Table of Contents:
Synopsis - Definition - Overview - Related Content
Synopsis
Congenital Muscular Dystrophy is a group of diseases of the muscles. Understanding and technology related to these diseases are progressing.
Topic Definition
- Congenital Muscular Dystrophies (CMDs)
Congenital muscular dystrophies (CMDs)are autosomal recessively-inherited muscle diseases. Congenital muscular dystrophy is one of the variants of muscle weakness disorders presenting early in life during infancy and soon after birth. Congenital muscular dystrophies are extremely rare. The difference between congenital myopathies and muscular dystrophies is that dystrophies are gradually progressive and are associated with increased muscle breakdown with age. Duchenne muscular dystrophy is the most common of all congenital muscular dystrophies. The diagnosis of CMDs requires the concurrence of expertise in multiple specialties, such as neurology, morphology, genetics, and neuroradiology, available in a few centers worldwide that have achieved sufficient experience with the different CMD subtypes.
Overview
Medical Overview
The congenital muscular dystrophies represent a group of muscle diseases. As technology and our understanding of these diseases progress, the CMDs are emerging from being a poorly understood subset of muscular dystrophy into a dizzying array of distinct diseases that share the onset of muscle weakness in infancy or childhood.
The U.S. Social Security Administration (SSA) has included Ullrich Congenital Muscular Dystrophy and Fukuyama Congenital Muscular Dystrophy as Compassionate Allowances to expedite a disability claim.
The only epidemiological study of the CMDs comes from a study in northern Italy, which placed disease prevalence at 8 x 106 (Mostacciulo ML et al., 1996). This suggests that CMD, though rare, as a group represents one of the more common neuromuscular disorders.
The best way to understand the CMDs is to follow a classification scheme proposed in a recent review article based on the location of the affected protein (Muntoni 2004, Mendell 2006). To better understand this classification, let us look at a model of the muscle cell membrane.
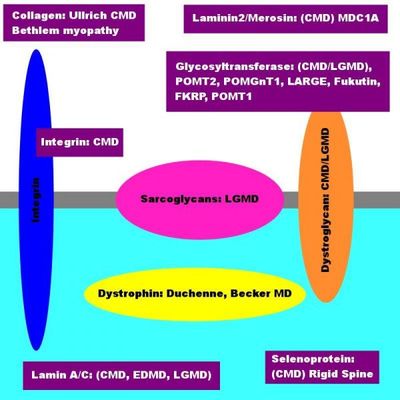
This image represents several things, including
The proteins which are involved in the various forms of muscular dystrophy.
- Yellow circle: The yellow circle illustrates the Dystrophin Proteins.
- Pink circle: The pink circle illustrates the Sarcoglycan Proteins.
- Orange circle: The orange circle illustrates the Dystroglycan Proteins.
- Blue circle: The blue circle illustrates the Integrin proteins.
- Purple boxes: The purple circle illustrates Collagen, Laminin Alpha2, Glycosyltransferase Enzymes (POMT1, POMT2, POMGnT1,
- FKRP, Fukutin and LARGE), Lamin A/C, and Selenoprotein).
The Dystrophin-Dystroglycan complex on the muscle cell membrane.
- White Backdrop: The white backdrop illustrates Extracellular Matrix (outside the muscle cell).
- Light Blue Backdrop: The light blue backdrop illustrates the Intracellular Matrix (inside the muscle cell).
- Grey Divider Line: The grey divider line illustrates the Sarcolemmal Muscle Cell Membrane.
CMD: Six Purple Boxes - These six purple boxes illustrate:
- Collagen (Ullrich)
- Laminin (Merosin Deficient CMD, MDC1A)
- Glycosyltransferases (POMT1, POMT2, POMGnT1, LARGE, FKRP, Fukutin)
- Integrin Alpha 7
- Selenoprotein
- Lamin A/C
The gray dividing line across the diagram is representative of the Muscle Cell Membrane, which is diving the inside of the cell (illustrated in light blue) from the outside of the cell (illustrated in white, also referred to as the 'Extracellular Matrix.'
The three circles in the image represent the 'Dystrophin-Glycoprotein Complex' that spans the Muscle Cell Membrane. A mutation in any single of these proteins accounts for some form of Muscular Dystrophy in the person affected.
On the inside of the cell, you will find a yellow circle called 'Dystrophin.' Should there be a mutation present in the gene encoding for this protein, the child affected has Duchenne or Becker's Muscular Dystrophy.
Within the muscle membrane are two additional groups of proteins, 'Sarcoglycans' and 'Dystroglycans.' The Sarcoglycans belong to a group of proteins that, when deficient, lead to Limb-Girdle Muscular Dystrophy.
The CMDs involve several proteins which can be found in four different locations in the cell:
1.) The Extracellular Matrix proteins(Collagen, Laminin, Integrin Alpha 7 and 9)
2.) The Glycosyltransferase proteins: These are proteins that place a sugar on the Alpha Dystroglycan (Dystroglycanopathies or Glycosyltransferase Deficiencies).
3.) Intracellular Protein Deficiency (Selenoprotein)
4.) Intranuclear Intermediate Filament Deficiency (Lamin A/C) and Nesprin 1
The CMDs potentially involve proteins that interact with Dystroglycan, illustrated by the orange circle, which represents two proteins that sit on top of each other in the muscle cell membrane; Alpha and Beta Dystroglycan. Alpha Dystroglycan is believed to play a role in cell signaling and the stability of the cell membrane. It also interacts with the Extracellular Matrix.
Extracellular Matrix Proteins (Collagen Six, Laminin Alpha 2, Integrin Alpha Seven and Nine):
A Collagen Six issue
The Extracellular Matrix forms the outside environment surrounding the muscle cell. The Extracellular Matrix performs critical functions by supporting muscle cell stability and regeneration while allowing muscle cells to adhere to the matrix. The Extracellular Matrix contains Collagen Six, composed of three strands: ' COL6A1,' COLCa2,' and, 'COL6A3.' Mutation in the gene for any of these strands may lead to either an abnormal, deficient or absent one. A mutation may lead to partial or complete deficiency as well. Both autosomal dominant and recessive mutations may cause Ullrich CMD. Autosomal dominant inheritance may present Bethlem Myopathy. There is a clinical spectrum that connects Bethlem and Ullrich; a theme repeated throughout CMD subtypes.
A mutation in any of these three genes may lead to Ullrich Congenital Muscular Dystrophy (UCMD). Bethlem Myopathy is a milder version in the spectrum of Collagen Myopathies. Persons with UCMD commonly experience muscle weakness, kyphoscoliosis, hip dislocation at birth, prominent heel bones, hyper-pigmented skin lesions, hyper-extensible finger joints, and elbow contractures. The fact that collagen is not present in the brain finds children with normal intelligence and no brain involvement. Respiratory issues may lead to the need for breathing assistance in the person's first or second decade. Following Laminin Deficiency, UCMD very likely represents the second most common form of CMD.
A Laminin (also referred to as 'Laminin Alpha 2' and 'Merosin') Issue
There are many forms of Laminin. Laminin-2 and Laminin-4 are particularly important to nerves and muscles. Laminin-2 is a protein part of the Basement Membrane that covers muscle cells, attaching to Alpha Dystroglycan and Integrin, then binding directly and indirectly to Collagen, Agrin, and additional molecules in the Extracellular Matrix. Laminin-2 plays roles in cell-to-cell recognition, cell differentiation, and cell survival. Mutations in Laminin-2 lead to Merosin Deficient CMD.
Children with mutations in Laminin-2 experience weakness and floppy tone; some require feeding and breathing support, depending on the severity of the illness. Many children do achieve the ability to sit unsupported, yet rarely achieve the ability to ambulate. They may develop contractures at the ankles, knees, hips, and elbows, followed by spinal rigidity and Scoliosis. MRI scans show white matter changes; the children affected have normal intelligence unless there is a structural brain abnormality - this can happen in a small percentage of children. Whether or not there is a structural brain abnormality present, children may experience seizure activity. Laminin is also present in peripheral nerves, leading to peripheral neuropathy.
Respiratory difficulty is one of the main complications of this disorder. Muscle weakness and spinal deformities may cause children to develop breathing difficulties during the night, limiting their ability to exchange oxygen for carbon dioxide effectively. Using non-invasive ventilation or bipap overnight can help overcome these difficulties. Difficulty with swallowing may require the placement of a gastronomy tube. Primary Laminin Deficiency may account for approximately thirty to forty percent of CMDs.
An Integrin Alpha Seven Issue
Integrin is a protein that spans the muscle cell membrane; Alpha Dystroglycan and Integrin can bind to Laminin-2. A primary deficiency in Integrin has been found in only three people who presented with delayed motor milestones and an abnormal muscle biopsy confirmed by genetic analysis to have a mutation in the Integrin gene.
An Integrin Alpha Nine Issue
Integrin Alpha Nine CMD has many similarities with the Collagen Six disorders, such as Ullrich and Bethlem. It is unclear whether Integrin Alpha Nine sits in the Extracellular Matrix or spans the Sarcolemmal Membrane (Integrin Alpha Seven). The genetic locus for Integrin Alpha Nine was initially reported in the French Canadian population because of an underlying founder mutation with subsequent gene identification as Integrin Alpha Nine.
Integrin Alpha Nine presents with distal extremity hyperlaxity, hypotonia, scoliosis, proximal contractures, normal to mildly elevated CPK, and normal intelligence. Some features differentiate Integrin Alpha Nine from Ullrich CMD, including a lack of high arched palate, a rigid spine, prominent calcaneus, and skin findings. Persons affected retain the ability to ambulate into the later decades of their lives, and while they demonstrate diminished respiratory capacity, they do not require ventilatory support.
The Glycosyltransferase Issue: The Glycosyltransferase Proteins affect Alpha Dystroglycan
Alpha Dystroglycan is coated with sugar molecules that must be attached through a complicated process involving several different genes. The following group of CMDs result from either partially functioning or missing enzymes whose job is to place a sugar on the Alpha Dystroglycan. Alpha Dystroglycan is found in many cells, including brain and muscle cells; this helps explain muscle cell weakness and brain involvement in CMDs. Many persons may also experience eye involvement.
Six genes have been isolated to date; more remains to be discovered. Each gene encodes a protein that places a particular form of sugar on Alpha Dystroglycan. The six genes are 'POMT1,' 'POMT2,' FKRP,' 'LARGE,' 'POMGnT1,' and 'Fukutin.' There is wide variability in disease despite the protein involved, based on the type of genetic mutation and the degree of functional protein. Children may show Microcephaly, structural brain abnormalities, adducted thumbs, mental retardation/delays, and seizures. Eye involvement may be a part of the presentation; retinal detachment or nearsightedness may be involved. Several children do not achieve the ability to ambulate. Muscle weakness and sometimes muscles that appear enlarged are present.
Historically, these diseases were named 'Walker Warburg Syndrome (WWS),' 'Fukuyama,' and 'Muscle-Eye Brain (MEB)' Disease; this was before the understanding of the genetic mutations underlying these diseases. The formerly Walker Warburg Syndrome represents the most severely affected children with CMD, which affects children under the age of three, causing both brain and eye abnormalities. Fukuyama CMD was first described in Japan in 1960, characterized by severe brain involvement, muscle weakness, mental retardation, seizures, and milder eye involvement. Children affected by Fukuyama CMD between the ages of two and eight may achieve the ability to stand up. Yet, progressive weakness develops with both respiratory and heart involvement in their second decade of life.
An Intracellular Protein (Selenoprotein N1 deficiency)
Selenoprotein N1 is an intracellular protein, meaning that it is inside the cell, located on the Endoplasmic Reticulum. A mutation in this gene leads to Rigid Spine Syndrome (RSMD1). Persons with RSMD1 experience difficulty walking due to thigh muscle weakness, spinal rigidity, and mild Achilles tendon tightness. Most children with RSMD1 achieve the ability to walk yet may develop Scoliosis as well as respiratory difficulty. Breathing difficulties may go unnoticed initially and may be experienced before difficulties with walking. Mutations in the same gene are responsible for congenital myopathy called 'Multi-Minicore Myopathy,' a name for a characteristic microscopic change in the muscle.
An Intranuclear Protein (Lamin A/C)
Lamin A/C is an Intranuclear Intermediate Filament attached to the Inner Nuclear Membrane. Mutations in Lamin A/C may lead to several different disorders, notably Emery Dreifuss Muscular Dystrophy, which may also include Cardiomyopathy with Conduction abnormalities. Recently, children with congenital onset of Lamin deficiency were found presenting with prominent weakness in the neck, known as 'Dropped-head Syndrome,' as well as spinal rigidity, scoliosis, and apparent weakness in Scapuloperoneal distribution. Muscle weakness and wasting are generalized yet particularly pronounced in their feet, arms, and neck.
Merosin Positive CMD
The term 'Merosin Positive CMD' refers to persons who fit a CMD picture with floppy muscle tone and may present with respiratory and feeding difficulty and whose muscle shows the presence of Merosin and a Dystrophic pattern. The exact genetic mutation and etiology have not been found. Merosin-positive CMD is a nonspecific diagnosis; because of this, persons with Merosin-positive CMD may have a variable presentation. A potential number of genes can account for this Merosin positive picture. It is hoped that new therapeutic targets will be identified by finding these genes and findings that will contribute to a greater understanding of Muscular Dystrophy as a whole.
Mutations found in two extended families with Merosin positive CMD mapped to Chromosome four. Family members presented with hypotonia at birth, without respiratory or feeding issues, normal to mildly elevated CK, and no contractures. The affected muscles included the trunk, shoulder, girdle, facial, neck, and proximal limb muscles without muscular hypertrophy. Their echocardiograms were normal; their intelligence was as well. Most of the affected family members had a stable course of muscle weakness; they achieved and maintained ambulation throughout adulthood.
 Author Credentials: Ian is the founder and Editor-in-Chief of Disabled World, a leading resource for news and information on disability issues. With a global perspective shaped by years of travel and lived experience, Ian is a committed proponent of the Social Model of Disability-a transformative framework developed by disabled activists in the 1970s that emphasizes dismantling societal barriers rather than focusing solely on individual impairments. His work reflects a deep commitment to disability rights, accessibility, and social inclusion. To learn more about Ian's background, expertise, and accomplishments, visit his full biography.
Author Credentials: Ian is the founder and Editor-in-Chief of Disabled World, a leading resource for news and information on disability issues. With a global perspective shaped by years of travel and lived experience, Ian is a committed proponent of the Social Model of Disability-a transformative framework developed by disabled activists in the 1970s that emphasizes dismantling societal barriers rather than focusing solely on individual impairments. His work reflects a deep commitment to disability rights, accessibility, and social inclusion. To learn more about Ian's background, expertise, and accomplishments, visit his full biography.