Cystic Fibrosis: Causes, Symptoms, Treatments
Author: Disabled World (DW)
Updated/Revised Date: 30 Dec 2025
Table of Contents:
Synopsis - About This Section - Publications - Subtopics
Synopsis
Information on Cystic Fibrosis, an inherited genetic disease involving the body mucus producing glands and the lungs.About This Section
Defining Cystic Fibrosis
Cystic fibrosis (CF), also known as mucoviscidosis, is a genetic disorder that affects mostly the lungs but also the pancreas, liver, kidneys, and intestine. Long-term issues include difficulty breathing and coughing up sputum as a result of frequent lung infections. Other symptoms include sinus infections, poor growth, fatty stool, clubbing of the finger and toes, and infertility in males among others. Different people may have different degrees of symptoms.
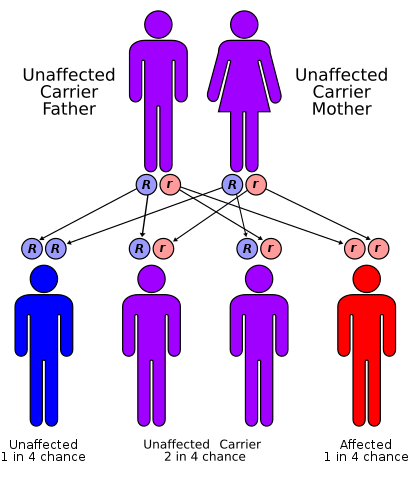
Cystic Fibrosis is a disease that is transmitted from both parents to a child if both parents carry the recessive gene, yet do not have the disease themselves. If both parents have this recessive gene and have a child, there is a twenty-five percent chance that the child will develop Cystic Fibrosis, a fifty-percent chance that the child will carry the gene but not develop Cystic Fibrosis, and a twenty-five percent chance that the child will not be affected in any way.
Approximately one in two-thousand five-hundred Caucasian children are affected by Cystic Fibrosis. More than ten million persons unknowingly carries the disease through the recessive gene, and bear no symptoms. Cystic Fibrosis is the most common hereditary cause of fatality in America.
Persons with Cystic Fibrosis commonly live between twenty-eight and thirty years, but some live years longer. Drainage procedures and medications are helping children with Cystic Fibrosis who would have died in the past to live into middle-adulthood and longer. The cause of death in persons with Cystic Fibrosis is usually either respiratory distress or a respiratory tract infection with enlargement of the right side of their heart.
Causes of Cystic Fibrosis
Researchers found a gene in 1989 called, 'Delta F508,' that causes Cystic Fibrosis. The gene is responsible for the disruption of a protein known as, 'Cystic Fibrosis Transmembrane Conductance Regulator (CFTR),' which causes the symptoms of Cystic Fibrosis. CFTR is produced within cells lining the glands in a person's small intestine, sweat glands, pancreas, and respiratory passages. It moves towards the cell's surface, where it then controls the flow of sodium in and out of the cell. For persons with Cystic Fibrosis, the CFTR protein is prevented from reaching the cell's outer surface, and salt (sodium) is prevented from exiting cells. The person's body attempts to compensate for this by overproducing secretions like mucus, sweat and water, which build up and cause the symptoms of Cystic Fibrosis.
Symptoms of Cystic Fibrosis
The symptoms related to Cystic Fibrosis vary among individuals with the disease. For some people, symptoms appear soon after they are born, while in other cases symptoms may not appear for months or even years. The symptoms of Cystic Fibrosis in infants and babies include pale stools, persistent diarrhea, foul smelling, bulky and greasy stools, gassiness, and abdominal swelling. Other symptoms include a chronic cough, frequent wheezing, pneumonia, or a chronic cough with thick mucus. Infants and babies with Cystic Fibrosis may also have vomiting, dehydration, salty-tasting skin, and intestinal blockages.

Children with Cystic Fibrosis experience symptoms including difficulty breathing, frequent respiratory infections, cough, rapid respirations, and a barrel-chested appearance. They may also experience fevers, flaring nostrils, and poor growth. Additional symptoms include poor appetite, malnutrition, gassiness, and abdominal discomfort and pain.
Cystic Fibrosis can present additional medical issues including both rounding and enlargement of the toes and fingers called, 'clubbing;' and Pneumothorax, a rupture of the lung tissues and trapping of air between the lungs and the chest. Cystic Fibrosis can also present sinus and nasal issues such as Nasal Polyps, a form of fleshy growth inside the nose, and Sinusitis, an inflammation of the nasal sinuses. Persons with Cystic Fibrosis may experience pancreatic, gallbladder, or liver problems, an enlargement of the right side of the heart, or cough up blood. They may experience delayed puberty, or reproductive abnormalities such as sterility in males. Over ninety-percent of males with Cystic Fibrosis are sterile.
Diagnosing
There are three stages at which Cystic Fibrosis can be determined; prenatal, postnatal, and during early childhood. The amniotic fluid surrounding a fetus in pregnant women is something that can be tested for fetal intestinal enzymes using a procedure called, 'Amniocentesis.' During the procedure, a sample of the amniotic fluid is extracted and analyzed for levels of intestinal enzymes; if the fetus has Cystic Fibrosis, these levels will be decreased.
There is a test available for newborns referred to as the 'Immunoreactive Trypsinogen Test (IRT), where a blood sample is drawn after the child is two or three days old. The sample is analyzed for Trypsinogen, which is a specific protein. In newborns with Cystic Fibrosis, Trypsinogen levels will be elevated.
For both children and young adults, there is a test available called the 'Electrolyte Sweat Test,' which measures the number of electrolytes in the person's sweat. The electrolytes measured include potassium, chloride, and sodium. The test is performed through application of a chemical called, 'Pilocarpine' to the person's forearm, and the use of a mild electric current to produce sweat. If higher than expected amounts of both chlorine and sodium are found in the test, the person has Cystic Fibrosis.
Other tests available which can help in the diagnosis of Cystic Fibrosis include Lung Function Tests, X-Rays, Stool Examinations, and Sputum (Phlegm) tests.
Genetic Testing
Family members including sisters, brothers, and first cousins in families with Cystic Fibrosis should all be tested to find out if they carry the gene responsible for the disease, as well as to find out if they have either digestive or lung problems.
Genetic testing for Cystic Fibrosis involves sampling of any form of tissue. Red or white blood cells found on the inside of a person's cheek, for example, can be analyzed for genes.
Treating Cystic Fibrosis
There is currently no cure for Cystic Fibrosis.
There are treatments for the symptoms of Cystic Fibrosis to include symptoms related to liver, digestive, lung, and gallbladder issues, as well as infertility.
Treatment of Lung Problems
Treatment of lung issues, such as inflammation, airway blockages, and bacterial infections, involves both medications and other methods. Antibiotics are used to treat lung infections; antibiotics such as Penicillins, Aminoglycosides, Cephalosporins, Ethambutol, Antimicrobial drugs, and Azeteonam. These antibiotics might be administered through injections, orally, or through inhalation in aerosol form.
Persons with Cystic Fibrosis may be prescribed Non-Steroidal Anti-Inflammatory Drugs (NSAID's) to treat inflammation. The NSAID's prescribed may include Prednisone or Ibuprofen, or Pentoxifylline, which have proven effective in reducing inflammation.
Airway blockages in persons with Cystic Fibrosis may involve treatment using Dornase Alfa, also known as, 'Pulmozyme.' Pulmozyme is administered through an inhalant machine called a nebulizer. A physician may advise a person with Cystic Fibrosis to perform Bronchial Drainage Chest Physiotherapy with Dornase Alfa treatment. Bronchial Drainage is done either mechanically or manually.
Manual chest Physical Therapy may be done in two ways. 'Autogenic Drainage,' is performed through using controlled breathing techniques which last approximately thirty to forty-five minutes. Another method involves placing the patient in a position which allows them to drain mucus from their lungs as their back or chest is both vibrated and clapped in or to dislodge mucus and assist in moving it out of the person's airways. The therapist repeats the process over different areas of the person's back and chest to loosen mucus in various areas of their lungs.
Mechanical treatment of airway clearance finds three devices available for therapy. One device is, 'Positive Expiratory Pressure Treatment,' which involves positive expiratory pressure treatment using a mouthpiece or face mask, which is attached to a one-way valve that has a set resistance of five to twenty centimeters of water. Another device is known as the 'Flutter,' which is a hand-held instrument resembling a pipe. There is a steel ball in the bowl of the pipe; during expiration it produces oscillations of variable breathing resistance. A third mechanical treatment involves an inflatable vest and high-frequency chest compression.
Persons with Cystic Fibrosis, who have not responded to treatment and have seriously reduced lung function, as well as a sharp decline in their health, may face a doctor recommended lung transplant. The three-year survival rate for persons with Cystic Fibrosis who have undergone a lung transplant is approximately fifty-five percent.
Treatment of Digestive Problems
Digestive problems associated with Cystic Fibrosis might be managed through a high-calorie, well-balanced diet which is high in protein and low in fat.
Vitamins A, D, E, and K, as well as Pancreatic Enzyme, may be prescribed by a doctor to ensure proper nutrition.
Treatment of Gallbladder and Liver Disease
Treatment of Gallbladder Disease in persons with Cystic Fibrosis involves a 'Laparoscopic Cholecystectomy,' which is a removal of the gallbladder. A surgeon creates a tiny incision in the person's navel, and removes the gallbladder in surgery.
Treatment of Liver Disease in persons with Cystic Fibrosis involves Oral Dissolution Therapy, which consists of consuming, 'Ursodeoxycholic Acid,' also known as, 'Actigall.' Actigall dissolves formations in the person's liver.
Treatment of Infertility
Men with CF can have their own biological children with the assistance of PESA, IVF and ICSI for their partner.
Women with Cystic Fibrosis may become pregnant through either natural means or intrauterine insemination. Pregnancy could present a threat to the life of a woman with Cystic Fibrosis who has a serious lung impairment.
Cystic Fibrosis Facts and Statistics
- In Canada, there are approximately 4,000 people with CF.
- Cystic fibrosis is diagnosed in males and females equally.
- In the United States, 1 in 3,500 children are born with CF.
- Ireland has the world's highest prevalence of cystic fibrosis, at 1:1353.
- Cystic fibrosis is often called mucovicidosis in other regions of the world.
- In the United States, approximately 30,000 individuals have CF; most are diagnosed by six months of age.
- Children with cystic fibrosis have very salty sweat, due to the abnormal protein manufactured by the cystic fibrosis gene.
- Approximately 1 in 25 people of European descent, and one in 30 of Caucasian Americans, is a carrier of a cystic fibrosis mutation.
- CF is not curable at this time, but with today's improved treatment, most people with CF can lead reasonably normal and productive lives.
- CF is an inherited condition. For a child to be born with CF, both parents must carry the CF gene. Carriers of the gene do not have any symptoms of the condition.
- Until the 1980s, most deaths from CF occurred in children and teenagers. Today, with improved treatments, some people who have CF are living into their forties, fifties, or older.
- Cystic fibrosis is most common in white people of Northern European ancestry, but also occurs in Hispanics, African-Americans and some Native Americans. It is rare in people of Asian and Middle Eastern origin.
- Today, more than one million Australians carry the faulty CF gene. One in every 25 people, frequently unknowingly, carries the CF gene. Some 80% of parents who have a child with CF were unaware they were carriers.
- Genetic carrier testing is available, and involves testing a sample of saliva or blood to see if you carry the gene for cystic fibrosis. This test may be offered to pregnant women, or trying to become pregnant, and their partners.
- In 1997, about 1 in 3,300 Caucasian children in the United States was born with cystic fibrosis. In contrast, only 1 in 15,000 African American children suffered from cystic fibrosis, and in Asian Americans the rate was even lower at 1 in 32,000.
- More than 1,000 different mutations in the CFTR gene have been identified in cystic fibrosis patients. The most common mutation (in 70% of cystic fibrosis patients) is a three-base deletion in the DNA sequence, causing an absence of a single amino acid in the protein product.
Curated and edited by Ian C. Langtree, Founder & Editor-in-Chief, Disabled World. This section is maintained by the Disabled World editorial team.
Last updated: